企画開発
医薬品のうち、動物を対象としたものを“動物用医薬品”と言いますが、一言に『対象が動物である』といっても、動物には様々な種類がありますし、また鶏、豚、牛、養殖水産動物といった経済動物や犬、猫のようなペット(愛玩動物・伴侶動物)までその位置付けも様々です。ですから、それぞれの対象動物に見合った医薬品の開発が必要となるのです。
また、動物用医薬品が人体用と異なる大きな点は、医薬品を使用した動物が最終的に食品となった場合の安全性が保証されなければならない事です。加えて、家畜や養殖水産動物といった産業動物を対象とする医薬品は、経済面に関しても有用でなければなりません。どんなに効果のある医薬品であっても、薬剤費に対して得られる経済効果が低いと、畜水産物の生産コストが上がってしまう為、現実的でない事もあります。動物用医薬品は、それ自体の安全性と有効性だけでなく、食品としての安全性、更には経済面における有用性が求められるという大変な課題を背負っています。
一般に、新薬の開発は、膨大な時間と金額を費やす事を御理解いただかなくてはなりません。私達の使命は、少しでも早く新薬を開発し、供給する事なのです。その為に、開発に要する時間の短縮や経費の捻出をするべく、会社同士の吸収・合併が繰り返されています。
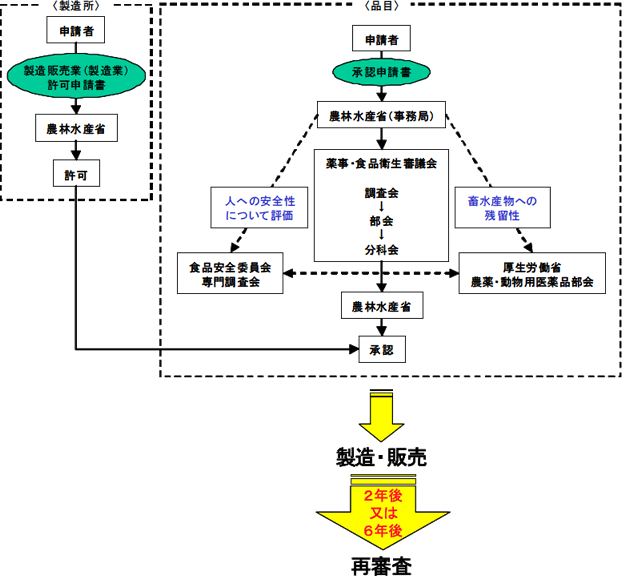
新薬の開発から供給に至るまでの流れは図1 に示す通りです。新薬を製造・販売する為には、国からの承認が必要となり、開発者あるいは申請者は種々のデータを記載した承認申請書を国に提出します。国からの承認は、品目毎に成分・分量、用法・用量、効能・効果、毒性、副作用、残留性等を、関係各分野の専門家で構成される薬事・食品衛生審議会で審議し、動物用医薬品承認の基準に照らし問題がないと認められたものだけに与えられます。
審議に必要な資料は、後述する農林水産大臣の定めるGLP 省令及びGCP 省令に従って収集され、作成されたものでなければなりません。
申請された品目が畜水産物に残留した場合に問題が生じる恐れのある場合は、厚生労働省においても残留に関する事項について審議を行います。また、人の健康への影響についても、内閣府の食品安全委員会で評価されます。
承認を受ける新薬は、同時に製造所(工場)についても国の許可(体外診断用医薬品の場合は登録)を得なければならず、国からの承認・許可(登録)がなされてはじめて製造・販売する事が可能となります。
図1 承認までの流れ(概要)
| 資 料 項 目 | 資 料 内 容 |
|---|---|
| 1. 起源又は発見(開発)の経緯等 | 起源又は開発の経緯、外国での使用状況等 |
| 2. 物理的・化学的試験資料 | 構造及び物理的・化学的・生物学的な性状に関する資料、規格及び検査方法設定資料等 |
| 3. 製造方法に関する試験資料 | 製造方法の設定の根拠となった試験資料等 |
| 4. 安定性試験資料 | 長期保存試験、苛酷試験(光、温度、湿度等の条件の相違による影響を経時的に見た試験)等の資料 |
|
|
|
|
対象動物について、通常投与量の最高量以上を投与又は使用し、安全性を確認した試験資料 |
7. 薬理試験資料
|
|
| 8. 吸収、分布、代謝及び排泄に関する試験資料 | 当該医薬品の血中濃度、尿・糞中排泄量、胆汁中排泄量、各臓器内濃度の経時的変化等に係る試験資 |
| 効能又は効果を裏付ける臨床試験資料 | |
|
|
食用に供される畜水産物への移行、残留に関する対象動物についての試験資料 |
GLP (Good Laboratory Practice:動物用医薬品の安全性に関する非臨床試験の実施の基準)
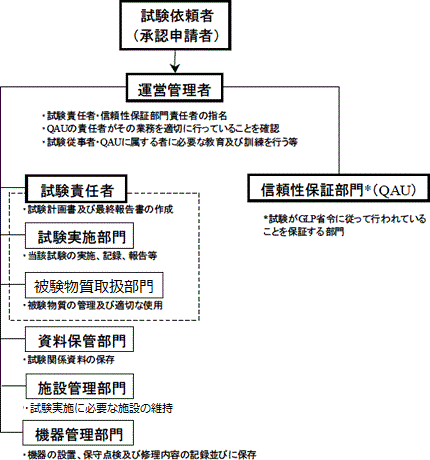
動物用医薬品等GLP とは、動物用医薬品等の承認申請書に添付される資料のうち、安全性に関する資料(毒性試験資料、対象動物に対する安全性試験資料及び残留性に関する試験資料)の信頼性を確保する為の基準を定めた省令(動物用医薬品:平成9 年10 月21 日農林水産省令第74 号、動物用医療機器:平成17年3月29日農林水産省令第31号、動物用再生医療等製品:平成26年11月18日農林水産省令第60号)です。本省令は、試験施設の運営管理、試験設備、試験計画、内部監査体制、信頼性保証体制、試験結果等について規定しています。
非臨床試験が適正に実施されたかどうかについては、内部監査を行うほか、農林水産省による実地調査が行われる場合もあります。
図2 非臨床試験実施体制(例)
GCP (Good Clinical Practice: 動物用医薬品の臨床試験の実施の基準)
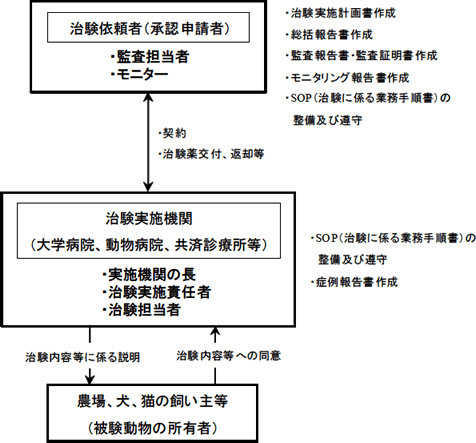
動物用医薬品等GCP とは、動物用医薬品等の承認申請書に添付される資料のうち、臨床試験に関する資料の信頼性を確保する為の基準を定めた省令(動物用医薬品:平成9 年10 月23 日農林水産省令第75 号、動物用医療機器:平成17年3月29日農林水産省令第32号、動物用再生医療等製品:平成26年11月18日農林水産省令第61号)です。本省令は、臨床試験が「倫理的な」配慮の下に、「科学的に」適正に実施されることを目的に制定されたものであり、臨床試験の実施に関する遵守事項を細かく規定しています。
「倫理的な」配慮としては、被験動物の所有者から治験に関する充分な情報を提供した上で同意を得る事、万一の事故が発生した場合の補償措置を講じる事、治験薬が残留している動物の肉、乳その他の生産物が食用に供される事のないよう措置を講じる事等について規定されています。
「科学的に」適正に試験を実施する為には、臨床試験に係る資料の収集・作成について、これら試験に関わるものが遵守すべき試験の計画、実施、モニタリング、監査、記録、報告等の事項が規定されています。
また、動物用医薬品等GLP と同様、臨床試験が適正に実施されたかどうかについて、農林水産省による実地調査が行われる場合もあります。
図3 臨床試験実施体制(例)
再審査・再評価
医薬品又は再生医療等製品は、医薬品医療機器等法に基づく承認審査を受け、承認を得られて初めて市販する事ができますが、限られた条件の中で設定された試験成績等を基に承認されている医薬品又は再生医療等施製品の効能・効果、性能や安全性を市販後にもう一度見直す為、「再審査」及び「再評価」の2 つの制度を設けており、この再審査、再評価に耐えないものは、承認を取り消される事があります。
医療機器又は体外診断用医薬品についても、医薬品医療機器等法に基づく承認審査を受け、承認を得られて初めて市販する事ができますが、医薬品又は再生医療等製品のような再審査・再評価の制度は適用せず、代わりに「使用成績評価」により対象品目が承認拒否事由に該当しないことを確認することになっています。
1.再審査(医薬品医療機器等法第14 条の4)
新規に承認された医薬品又は再生医療等製品(新医薬品等)について、医薬品医療機器等法で承認後、一定期間内に承認の見直しをする制度です。
新医薬品等の承認に際しては、従来から詳細な資料の提出が求められ、厳格な審査が実施されてはいますが、承認時までのデータでは、特に、臨床試験成績において症例数、使用範囲等におのずと制約があります。再審査とは、この問題点を解消する為、承認後も引き続き新医薬品等の使用成績等の調査を行い、原則として6 年後(但し、既承認動物用医薬品と有効成分、投与経路が同一であるもので、その効能・効果もしくは用量の異なる医薬品については2 年後。再生医療等製品は農林水産大臣から指示された期間。)にその新医薬品等の効能・効果や安全性等の再確認をする、という制度です。
その為、新医薬品等の承認を取得した者(これを製造販売業者と言います。)は、通常、承認後6 年間にわたって新医薬品等の野外における調査を実施する事とされています。効能・効果については、市販後の使用成績及び文献調査等の資料等、安全性については、市販後の副作用情報及び文献調査等の資料をまとめ、農林水産大臣に報告します。
これに基づき、新医薬品等の有効性及び安全性を見直す審議が行われ、その結果によっては、新たに承認の見直しを求められたり、承認の取り消しを求められたりする事もあります。
なお、再審査の為の申請資料は、GPSP*3)省令(Good Post-Marketing Study Practice, 動物用医薬品の製造販売後の調査及び試験の実施の基準に関する省令(平成17 年3 月29 日農林水産省令第33号)、動物用再生医療等製品の製造販売後の調査及び試験の実施の基準に関する省令(平成26年11月25日農林水産省令第63号)に適合しなければなりません。この省令は、再審査資料作成の為の調査又は試験を適正かつ円滑に実施する事により、その信頼性を確保する為の基準です。これにより、仮に有効性又は安全性が疑わしい結果が得られた場合においても、その調査結果及び試験成績等について適正に検討及び評価が行われ、新医薬品等の安全対策の充実強化も図られることとなります。つまり、製造販売業者が新医薬品等の承認・販売を維持する為に有益な調査結果や試験成績のみを再審査申請の資料として記載する事ができない制度となっています。
2.再評価(医薬品医療機器等法第14 条の6)
再審査における審議が終了した医薬品等は、そのままでは、その後の有効性や安全性についての見直しがされず、長期にわたって使用される事になります。そこで、長い間使用されてきた医薬品等について、最新の獣医学、薬学等の科学的知見に基づき、品質、有効性及び安全性について見直しをするのが再評価制度です。
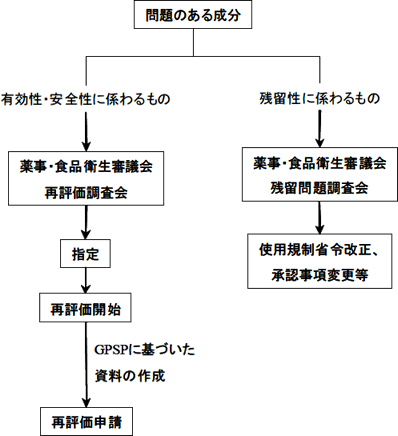
再評価の対象となるのは医薬品の主剤(有効成分)であり、農林水産大臣の公示により指定されます。
具体的には、農林水産省におけるスクリーニング(文献等の情報を広く収集、評価し、有効性、安全性について検討を要する情報をピックアップする事)により得られた情報をもとに、薬事・食品衛生審議会の審議を経て、再評価対象成分が指定される事となります。(図4 参照)図4 再評価制度の流れ(概要)
3.医療機器又は体外診断用医薬品の使用成績評価(医薬品医療機器等法第23条の2の9)
製造販売後も使用成績に係る調査を行い、一定期間後にその安全性等を再確認する必要があると判断される医療機器又は体外診断用医薬品(医療機器等)について、薬事・食品衛生審議会の意見を聴いて使用成績評価の対象として指定されます。新医療機器については、従来は承認後、法律で定められた期間に収集された使用成績に関する資料等に基づき、再審査を行い、有効性、安全性を確認してきましたが、平成25年の法改正により、新医療機器を一律に評価対象とするのではなく、医療機器の特性に応じて、必要な品目に一定期間の評価をすることで、必要な有効性及び安全性に関する情報が集約できるよう見直されました。また、新医療機器以外の医療機器及び体外診断用医薬品に該当する場合であって、品質、有効性及び安全性の確認がなされているものの、臨床試験データ等により重大な不具合が生じる可能性が懸念されているなど、特に使用成績の確認が必要と判断される場合も使用成績評価の対象となります。
使用成績評価の対象に指定された医療機器等の製造販売業者は、農林水産大臣の指定する期間、医療機器等の不具合の発生、不具合によるものと疑われる疾病、傷害若しくは死亡又はその使用によるものと疑われる感染症その他の使用成績等について調査を行います。調査結果の報告は、承認を受けた日から起算して1年ごとに年次報告を行い、農林水産大臣が指示する期間を経過して3か月以内の期間内に使用成績評価申請をして、農林水産大臣の使用成績に関する評価を受けることになります。使用成績評価の品性に係る効果又は性能及び安全性についての調査資料は、GPSP基準、GLP及びGCPの基準と同程度の信頼性の基準に適合されたものでなければなりません。評価の結果、有用性が認められない場合は、承認が取り消されることがあります。
再評価には、下記の通り、定期的な再評価と臨時の再評価の2 種類があります。
- ①定期的な再評価
- 承認、再審査、再評価その他農林水産省による評価が行われた時点から5 年が経過した成分について見直しを行うもの。見直しは、原則として農林水産省が入手した文献を元に行い、必要に応じ関係企業の意見が求められる。見直しを行った成分については、見直し後原則10 年が経過した時点で再度見直しを行い、以下これを繰り返す。
- ②臨時の再評価
- ① の定期的な再評価のほか、臨時に再評価を行う必要があると思われる場合においては、薬事・食品衛生審議会の審議を経て、再評価を行う必要がある成分として指定を行う。
再評価にかかる審議の結果、医薬品等としての欠格事由が確認された場合は、承認の取り消し又は承認事項の一部変更の手続をとることとなります。
なお、再評価の為の申請資料は、再審査と同様、GPSP 省令に適合しなければなりません。